






Researchers have identified a potential dual-pronged approach to treating Niemann-Pick type C (NPC) disease, a rare but devastating genetic disorder.
Researchers have identified a potential dual-pronged approach to treating Niemann-Pick type C (NPC) disease, a rare but devastating genetic disorder.
The research, based on studies of human nerve and liver cells grown from patient-derived induced pluripotent stem cells (iPSCs), is co-authored by Gates Cambridge Alumnus Sovan Sarkar and published in the journal Stem Cell Reports.
According to the US National Institutes of Health (NIH), approximately 1 in 150,000 children born are afflicted with NPC, the most common variant of Niemann-Pick. Children with NPC experience abnormal accumulation of cholesterol in their liver and nerve cells, leading to liver failure, neurodegeneration, and – ultimately – death, often before age 10. Although there is currently no effective treatment for NPC disease, a clinical trial examining potential cholesterol-lowering effects of the drug cyclodextrin in NPC patients is ongoing. The research, conducted at Professor Rudolf Jaenisch’s laboratory at MIT’s Whitehead Institute where Sovan is currently based, suggests that the high doses may actually be harmful.
“At those levels of cyclodextrin (in the clinical trial), Maetzel and her coauthors show that cells encounter a further block in autophagy that could be detrimental,” says Professor Jaenisch. “But when they use it at a lower dose in combination with another small molecule, carbamazepine, which stimulates autophagy, then it significantly improves the survival of the cells. Such an approach lowers cholesterol levels and restores the autophagy defects at the same time. This could be a new type of treatment for NPC disease.”
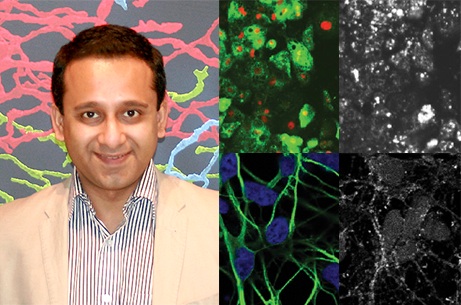
To clarify what is amiss in NPC disease and identify potential therapeutics that could correct these problems, Sovan’s co-author Dorothea Maetzel generated iPSCs from patients with some of the most common genetic mutations that cause NPC1. The iPSCs were created by pushing skin cells donated by the patients back to an embryonic stem cell-like state. These iPSCs were then differentiated into liver and neuronal cells, the cell types most affected in NPC1 disease. Additionally, one copy of a causal mutation was corrected by genome editing, in the NPC1 gene, to create control cells whose genomes differ only at the single edited gene copy.
When Dorothea and Sovan analysed the cellular functions in the NPC1-mutant and control cell lines, they determined that although cholesterol does build up in the NPC1-mutant cells, a more significant problem is defective autophagy – a basic cellular function that degrades and recycles unneeded or faulty molecules, components, or organelles in a cell. The impaired autophagy prevents normal elimination of its cargo, such as damaged organelles or other substrates like p62, which then accumulates and damages the cells. Previously, recent work by Sovan in the Jaenisch laboratory with cells from NPC1 disease mouse models has revealed that defective autophagy is a major causative factor for the disease pathology.
“Autophagy dysfunction has major implications in several neurodegenerative and certain liver conditions, and therefore autophagy modulators have tremendous biomedical relevance in these pathological scenarios, particularly for neurodegeneration”, says Sovan [2002], who did a PhD at Cambridge in Medical Genetics. “We wanted to screen for compounds stimulating autophagy in human disease-relevant cells and show the beneficial effects of such an approach in the context of a lipid/lysosomal storage disorder.”
The researchers used the two types of human disease-affected cells to screen for compounds known to improve autophagy but not impacting on the mammalian target of rapamycin (mTOR) pathway, which has critical cellular functions and also controls autophagy. They found only one capable of jumpstarting autophagy independently of mTOR in both liver and nerve cells. When this drug, carbamazepine, which is a mood stabiliser prescribed for bipolar disorder, was added in combination with low doses of cyclodextrin, both cholesterol accumulation and autophagy defects were rescued in the NPC1-mutated cells. Dorothea and Sovan say the next step is for the combination of drugs to be tested on animals. They add that they believe this cell type-specific screening approach can have broad relevance for other human diseases.
“In principle, we can extend the knowledge of some of our findings in human disease-relevant cells to related neurodegenerative and lysosomal storage disorders,” says Sovan. “I’m excited about the prospects of using patient-specific iPSCs to model diseases for mechanistic insights and drug screening that provides a physiological setting close to the disease context.” Sovan recently featured in the April 2014 issue of the journal Autophagy for his contribution to the field.
*Images show human hepatic (top panel) and neuronal (bottom panel) cells derived from NPC1 patient-specific iPSCs where accumulation of cholesterol was observed with Filipin staining (grayscale panels, right). Cellular images courtesy of Stem Cell Reports.




